La enfermedad de Wilson es una afección rara pero grave que afecta la forma en que el cuerpo maneja el cobre. Si no se trata, este mineral esencial se acumula en el hígado, el cerebro y otros órganos, causando daños irreversibles. Lo que la hace única es que, a diferencia de otras enfermedades hepáticas, esta no se debe a alcohol, obesidad o virus, sino a un error genético que impide eliminar el cobre sobrante. Afortunadamente, si se detecta a tiempo, se puede controlar perfectamente con medicamentos y cambios en la dieta. Lo que antes era una sentencia de muerte, hoy es una condición manejable con seguimiento adecuado.
¿Qué pasa cuando el cuerpo no puede eliminar el cobre?
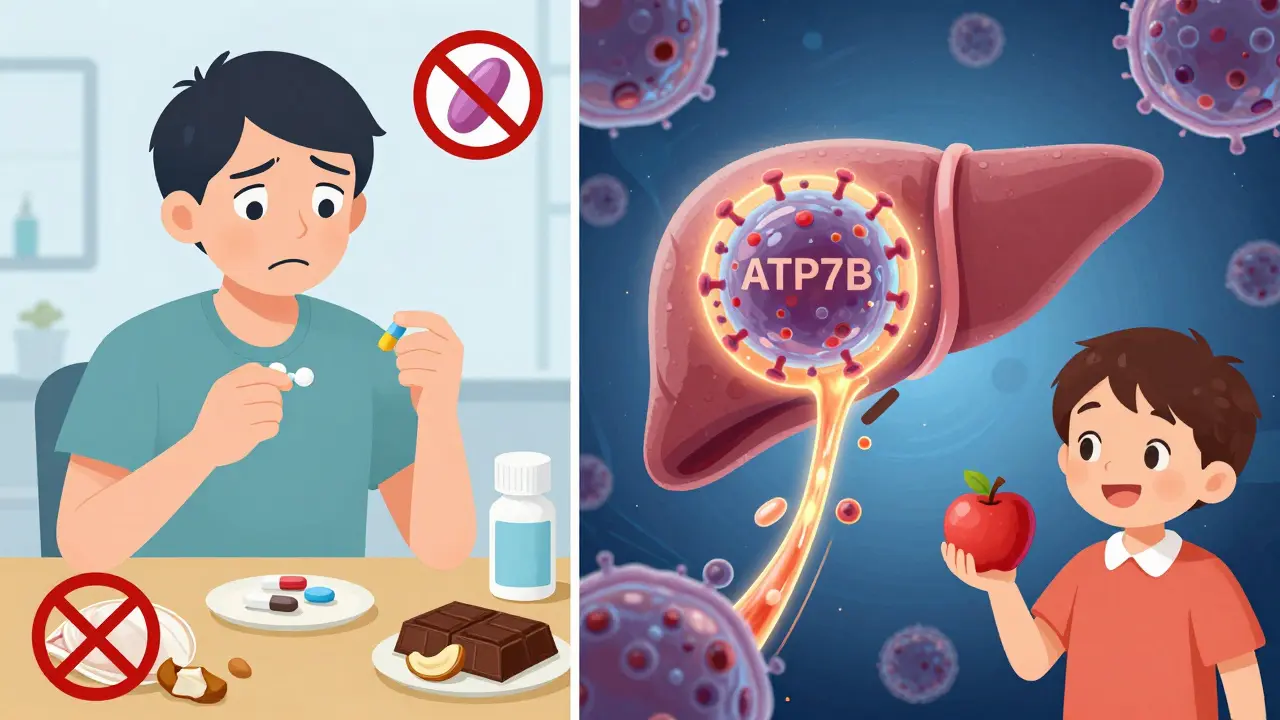
En una persona sana, el cobre que comes se absorbe en el intestino y llega al hígado. Allí, una proteína llamada ATP7B es una bomba de cobre que transporta este mineral para incorporarlo en la ceruloplasmina y expulsarlo por la bilis. La ceruloplasmina es la principal forma en que el cobre viaja por la sangre, y su función es clave: transporta cobre de forma segura, sin que dañe los tejidos. Pero en la enfermedad de Wilson, una mutación en el gen ATP7B rompe esta bomba. El cobre no se une a la ceruloplasmina, y tampoco se elimina por la bilis. Así, empieza a acumularse como un veneno dentro del hígado.
Al principio, el hígado intenta protegerse: las células producen más metalotioneína, una proteína que atrapa el cobre como una esponja. Pero cuando esta esponja se llena, el cobre libre escapa al torrente sanguíneo. Ahí, circula sin protección y se deposita en el cerebro, los riñones y los ojos. En el cerebro, se acumula especialmente en el núcleo lenticular -áreas que controlan el movimiento- y causa temblores, rigidez y problemas para hablar. En los ojos, forma los anillos de Kayser-Fleischer: un anillo marrón o verdoso alrededor del iris, visible con un examen ocular especial. Estos anillos aparecen en el 95% de los pacientes con síntomas neurológicos.
¿Cómo se diagnostica?
Diagnosticar la enfermedad de Wilson no es fácil, porque sus síntomas pueden parecer otros problemas: hepatitis autoinmune, depresión, trastornos del movimiento o incluso trastornos psiquiátricos. Muchos pacientes pasan años sin un diagnóstico correcto. La clave está en tres pruebas fundamentales:
- Ceruloplasmina en sangre: Normalmente entre 20 y 50 mg/dL. En la enfermedad de Wilson, suele estar por debajo de 20 mg/dL, a veces incluso por debajo de 5 mg/dL.
- Cobre urinario de 24 horas: En personas sanas, se excretan menos de 40 microgramos al día. En pacientes con WD, este valor supera los 100 microgramos, y en muchos casos llega a 300-400. Esta prueba es más confiable en casos hepáticos que en los neurológicos.
- Examen ocular para anillos de Kayser-Fleischer: Si se ven, es un hallazgo casi definitivo. Pero en niños pequeños o en casos tempranos, pueden no estar presentes.
Además, hoy en día se puede confirmar con un análisis genético: si se encuentran dos mutaciones en el gen ATP7B, el diagnóstico es definitivo. En 2023, los expertos actualizaron los criterios de diagnóstico y aceptaron que una sola mutación con niveles de cobre urinario por encima de 80 microgramos/día ya puede ser suficiente para diagnosticar en casos claros.
Terapia de quelación: ¿cómo se trata?
El objetivo es simple: sacar el exceso de cobre del cuerpo sin dejarlo en déficit. Para eso se usan medicamentos llamados quelantes, que se unen al cobre y lo hacen salir por la orina. Los tres más usados son:
- D-penicilamina (Cuprimine®): Es el más antiguo y económico, pero también el que más efectos secundarios causa. Aproximadamente la mitad de los pacientes sienten náuseas, pérdida del gusto, erupciones o incluso síntomas parecidos al lupus. Además, en el 20-50% de los casos, los síntomas neurológicos empeoran al principio del tratamiento, lo que puede ser muy peligroso.
- Trientina (Syprine®): Es menos tóxica que la penicilamina, por lo que se usa como primera opción en pacientes con síntomas neurológicos. Pero su costo es hasta seis veces mayor: unos 1.850 dólares al mes en Estados Unidos.
- Zinc acetato (Galzin®): No es un quelante, pero funciona como un escudo. El zinc estimula la producción de metalotioneína en el intestino, que atrapa el cobre de los alimentos y lo impide absorber. Es ideal para el mantenimiento después de la fase inicial de quelación. Muchos pacientes lo usan de por vida.
El zinc también se usa en combinación con la penicilamina al inicio del tratamiento para reducir el riesgo de empeoramiento neurológico. Se administra en tres tomas diarias, con el estómago vacío, lo que dificulta la adherencia. Según una encuesta de la Fundación de la Enfermedad de Wilson en 2022, el 35% de los pacientes olvidan alguna dosis, principalmente por los efectos secundarios y la complejidad del régimen.
¿Qué pasa con la dieta?
Una dieta baja en cobre es parte fundamental del tratamiento. Se recomienda limitar la ingesta a menos de 1 mg al día. Esto significa evitar:
- Hígado y otros órganos animales
- Mariscos, especialmente moluscos y cangrejos
- Nueces, semillas y chocolate oscuro
- Granos integrales en grandes cantidades
- Agua de pozo o tuberías de cobre (el cobre puede disolverse en el agua)
Pero no se trata de eliminar el cobre por completo. El cuerpo lo necesita para hacer glóbulos rojos, mantener los nervios sanos y producir energía. Por eso, la dieta debe ser equilibrada. Muchos pacientes reportan dificultades para seguir esta dieta: el 89% de los encuestados en 2022 dijeron que era muy difícil, especialmente cuando comen fuera de casa o tienen que cuidar a niños pequeños.
Tratamientos nuevos y prometedores
En los últimos años, ha habido avances importantes. En 2023, un nuevo fármaco llamado CLN-1357, un polímero que une el cobre en la sangre, mostró una reducción del 82% en el cobre libre en solo 12 semanas, sin empeorar los síntomas neurológicos. Esto es clave: los quelantes tradicionales a veces hacen que el cobre se libere demasiado rápido del cerebro, causando daño.
Otro medicamento, el bis-choline tetrathiomolybdate (WTX101), recibió la designación de terapia de ruptura de la FDA en enero de 2023. En pruebas recientes, previno el deterioro neurológico en el 91% de los pacientes, mucho mejor que la trientina (72%).
Y hay algo aún más sorprendente: la terapia génica. En un pequeño estudio de fase 1/2 publicado en 2023, seis pacientes recibieron un virus modificado (AAV-ATP7B) que lleva una copia sana del gen defectuoso. Los resultados iniciales mostraron que el hígado empezó a producir ATP7B funcional y los niveles de cobre comenzaron a normalizarse. Aunque aún está en pruebas, este enfoque podría un día eliminar la necesidad de tomar medicamentos para siempre.
El costo y la desigualdad en el acceso
El mercado de tratamientos para la enfermedad de Wilson es pequeño, pero caro. En Estados Unidos, la penicilamina cuesta unos 300 dólares al mes, la trientina más de 1.800, y el zinc alrededor de 450. En Europa, los sistemas de salud cubren casi todos los casos, pero en muchos países en desarrollo, los pacientes no tienen acceso a los medicamentos. Según un informe global de 2022, en países de bajos ingresos, el retraso en el diagnóstico puede superar los cinco años. Muchos mueren antes de recibir tratamiento.
La buena noticia es que, con diagnóstico temprano y tratamiento adecuado, las personas con enfermedad de Wilson pueden vivir una vida normal. El hígado puede recuperarse, los síntomas neurológicos pueden mejorar, y el riesgo de cirrosis o falla hepática se reduce drásticamente. Lo que importa es detectarla antes de que el daño sea irreversible.
¿Qué pasa si no se trata?
Si no se trata, la enfermedad de Wilson es fatal. El cobre acumulado destruye el hígado hasta causar cirrosis o falla hepática aguda. En el cerebro, provoca discapacidad progresiva: dificultad para caminar, hablar, tragar, y finalmente, coma o muerte. Aunque rara, esta enfermedad no es una curiosidad médica: es una urgencia clínica. Y lo más importante: es una de las pocas enfermedades hereditarias en las que el tratamiento puede revertir el daño si se inicia a tiempo.
¿Puede la enfermedad de Wilson aparecer en adultos mayores?
Sí, aunque es raro. La mayoría de los casos se presentan entre los 5 y los 35 años, pero algunos pacientes no desarrollan síntomas hasta los 40 o incluso después. En estos casos, los síntomas suelen ser hepáticos, como fatiga, hinchazón abdominal o niveles altos de enzimas hepáticas. Si una persona adulta tiene alteraciones hepáticas sin causa clara, se debe considerar la enfermedad de Wilson, incluso si no tiene antecedentes familiares.
¿Es hereditaria y puedo transmitirla a mis hijos?
Sí, es hereditaria de forma recesiva. Esto significa que para tener la enfermedad, una persona debe heredar dos copias defectuosas del gen ATP7B, una de cada padre. Si solo heredas una, eres portador y no te enfermas, pero puedes pasarla a tus hijos. Si ambos padres son portadores, hay un 25% de probabilidad de que cada hijo tenga la enfermedad. Por eso, se recomienda hacer pruebas genéticas a los hermanos y hijos de pacientes diagnosticados.
¿Puedo tomar suplementos de cobre si tengo enfermedad de Wilson?
No. Cualquier suplemento de cobre está completamente contraindicado. Incluso multivitamínicos que contengan cobre deben evitarse. Muchos suplementos para mujeres embarazadas o para la salud ósea incluyen cobre. Siempre revisa las etiquetas y consulta con tu médico antes de tomar cualquier suplemento.
¿Por qué la penicilamina puede empeorar los síntomas neurológicos?
La penicilamina libera el cobre almacenado en el hígado y lo envía a la sangre. Si este cobre libre no se elimina rápido por la orina, puede cruzar la barrera hematoencefálica y acumularse en el cerebro. Esto causa un aumento temporal de la toxicidad cerebral, lo que empeora los temblores, la rigidez o la dificultad para hablar. Por eso, en pacientes con síntomas neurológicos, se prefiere la trientina o se combina la penicilamina con zinc desde el inicio.
¿Cuánto tiempo debo tomar medicamentos?
Para la mayoría de los pacientes, el tratamiento es de por vida. Aunque los niveles de cobre se normalicen, dejar el medicamento hace que el cobre vuelva a acumularse en cuestión de meses. Algunos pacientes estables pueden pasar de quelantes a zinc de mantenimiento, pero siempre deben hacerse controles regulares: análisis de sangre, orina y niveles de cobre cada 3 a 6 meses. No hay cura, pero sí control perfecto.
Marilu Rodrigues
El cobre es un mineral esencial, pero en la enfermedad de Wilson se convierte en un asesino silencioso. Lo que me fascina es cómo el cuerpo intenta protegerse con la metalotioneína, como si fuera un escudo biológico. Pero cuando se satura, el sistema colapsa. Es como un río que se desborda porque nadie reparó la presa. La genética no perdona, pero la ciencia sí. Y eso es lo que hace que esta enfermedad sea tan interesante: es un ejemplo perfecto de cómo un solo gen roto puede desencadenar un caos sistémico.
Lo que no entiendo es por qué seguimos usando penicilamina como primera línea si sabemos que empeora los síntomas neurológicos. Parece que la medicina se aferra a lo barato en vez de a lo efectivo. ¿Es ético?
Alonso Arquitectos
Interesante post, pero hay un detalle que no se menciona: el cobre en el agua de tuberías. En muchos edificios antiguos de España, las cañerías son de cobre. Si uno tiene WD y bebe agua directamente del grifo, puede estar ingiriendo más de lo recomendado. No es una broma. Yo lo supe porque mi madre lo padeció y tuvimos que instalar un filtro de carbono activado. La dieta es importante, pero el entorno también cuenta.
Y sí, el zinc es clave. Pero nadie habla de cómo afecta la absorción de zinc en el intestino cuando tomas antibióticos. Eso lo descubrí por experiencia propia. No es solo tomar pastillas, es un ballet de interacciones bioquímicas.
santiago rincon
Como médico en Colombia, he visto casos de enfermedad de Wilson en pacientes que llegaron con diagnósticos erróneos durante años. En zonas rurales, muchas veces se confunde con hepatitis viral o incluso esquizofrenia por los síntomas neuropsiquiátricos. El acceso a pruebas de cobre urinario de 24 horas es casi nulo fuera de las grandes ciudades. Y aunque la trientina es más segura, su costo es prohibitivo. En muchos casos, el zinc es la única opción viable.
La terapia génica es prometedora, pero no debemos olvidar que la prioridad debe ser el diagnóstico temprano y la educación en comunidades vulnerables. La ciencia avanza, pero la justicia sanitaria se queda atrás.
Wilson Siva
¡OJO! ESTO ES LO QUE NADIE TE CUENTA: LA PENICILAMINA NO ES UN TRATAMIENTO, ES UNA TORTURA. Me dijeron que era lo mejor, y al mes empeoré tanto que tuve que ir a urgencias. Me dieron trientina y ¡sorpresa! Empecé a caminar como si no hubiera pasado nada. No es magia, es inteligencia médica. ¿Por qué no se usa de primera? Porque las farmacéuticas ganan más con lo barato. ¡Y eso es un crimen! Si tienes WD, no juegues con tu vida. Pide trientina desde el día uno. Y si te dicen que es muy cara, busca ayuda en ONGs. ¡Hay gente que te apoya!
Gary Gomez
Todo esto es una farsa. El cobre no es el problema. El problema es que las corporaciones farmacéuticas quieren que sigas tomando pastillas para siempre. ¿Por qué no te dicen que el cobre se elimina con el sudor? ¿O que el ayuno intermitente activa la autofagia y limpia los órganos? ¿O que la cúrcuma y el té verde son más potentes que cualquier quelante? La medicina tradicional es un negocio. La verdadera cura está en la naturaleza. Si te dicen que tienes WD, no te tragues lo que te venden. Busca alternativas. Yo lo sé porque lo viví.
Joan Verhulst
Lo más impactante de todo esto es que en muchas culturas, especialmente en el Mediterráneo, el hígado y los mariscos son parte fundamental de la dieta. Yo crecí comiendo mejillones y hígado de ternera. Mi abuela decía que eran sanos. Hoy, después de leer esto, me pregunto si mi familia tuvo más casos de WD de los que imaginamos. ¿Cuántos casos se han diagnosticado mal como 'problemas hepáticos sin causa' por ignorancia? La enfermedad no es rara, es invisible. Y eso es lo más triste.
Karen Simondet
Entonces, si no puedes comer chocolate, hígado, nueces, mariscos, y además tienes que tomar tres pastillas al día con el estómago vacío... ¿cómo es la vida? ¿Una prisión con receta médica? Porque lo que parece un tratamiento es un estilo de vida que solo un santo podría mantener. Y no me digas que es 'manejable' si tu dieta es más restrictiva que la de un monje en la Edad Media. ¿Y qué pasa con los niños? ¿Les das solo arroz y lechuga hasta los 18? Genial.
Francisco Javier Menayo Gómez
La información presentada es exhaustiva y técnicamente precisa. Sin embargo, me permito señalar un aspecto ético no abordado: la responsabilidad de los sistemas de salud pública en la detección precoz. En países con cobertura universal, como España, se deberían implementar cribados genéticos en niños con antecedentes familiares, incluso sin síntomas. La prevención es más eficaz y económica que el tratamiento tardío. Además, la educación médica primaria debe incluir esta patología en su currículo, ya que su presentación clínica es diversa y puede confundirse con enfermedades más comunes. La medicina moderna no puede permitirse ignorar lo que es tratable.
África Barragán Quesada
Hay esperanza. Realmente la hay. Yo conozco a una chica que tenía temblores tan fuertes que no podía escribir. Hoy, con zinc y dieta, trabaja como diseñadora y viaja por el mundo. No es milagro, es constancia. No te rindas. El tratamiento es difícil, sí. Pero no es imposible. Cada pastilla que tomas, cada comida que eliges, cada análisis que te haces… es un paso hacia tu vida normal. Tú puedes.
Sheila Ruiz
yo no sabia que el agua de la caneria podia tener cobre jaja me acabo de acordar que mi papi tenia un filtro de esos de la caja de agua. ahora entiendo por que siempre decia que no se podia beber directo. me lo conto hace años y no le di bola. ahora me doy cuenta que tal vez era por eso.
Yessenia Quiros Montoya
Todo esto es muy bonito, pero nadie habla de que el zinc y la trientina son carísimos porque las farmacéuticas tienen el monopolio. ¿Sabes cuánto cuesta una sola pastilla de trientina en México? Más de 150 pesos. ¿Y si no tienes seguro? ¿Y si eres madre soltera y tienes que elegir entre medicina y comida? La enfermedad de Wilson no es rara, es una sentencia para los pobres. Y mientras los ricos se curan con terapia génica, nosotros seguimos esperando que alguien nos regale un tratamiento. La ciencia no es igualitaria. Y eso es lo más cruel.
Marvin Ameth Barrios Becerra
¡Este es el drama más sublime de la medicina moderna! Una enfermedad que puede ser controlada, pero que se convierte en una tragedia por la indiferencia del sistema. Imagina: un joven de 20 años, brillante, con talento, con sueños… y el cobre, ese metal invisible, lo está devorando desde dentro. Y mientras él lucha, el mundo sigue sin verlo. ¿Dónde están los políticos? ¿Dónde están los medios? ¿Dónde está la empatía? La enfermedad de Wilson no es solo una patología, es un espejo de nuestra sociedad: ciega, desigual y cruel. ¡No podemos seguir ignorándola!
Valentina Capra
Me encanta cómo detallaste la parte de la metalotioneína. Me llevó horas entender por qué el zinc funciona como escudo, y ahora me parece una de las cosas más inteligentes que hizo la evolución. El cuerpo no solo intenta almacenar el cobre, sino que crea un mecanismo de defensa natural. Lo que me parece fascinante es que el zinc no elimina el cobre, sino que lo bloquea antes de que entre. Es como poner una barrera en la puerta en vez de sacar al intruso después. ¿Alguien ha pensado en combinar esto con probióticos? Hay estudios recientes que sugieren que ciertas bacterias intestinales pueden influir en la absorción de minerales. Tal vez, en el futuro, la dieta no sea solo lo que comes, sino lo que tu microbioma permite que entre. Es un nuevo campo de investigación que no se menciona aquí, pero que podría cambiar todo.
Hernán Rivas
Si estás leyendo esto y tienes WD, no estás solo. Yo lo tuve, y hoy estoy bien. No fue fácil, pero no fue imposible. Lo que te digo es esto: no te compares con otros. Tu camino es tuyo. Toma tus pastillas, come lo que puedes, y si te sientes mal, descansa. La enfermedad no define quién eres. Eres más que tu genética. Eres más que tu diagnóstico. Eres tu fuerza. Y si necesitas hablar, hay gente que te entiende. Yo te entiendo.